So you have the results of a GWAS analysis and want to see if any specific pathways are enriched. This is a summary of how you could do that using one of a number of tools available, the SNP ratio test (SRT) http://snpratiotest.sourceforge.net/
This assumes you have the data in PLINK format.
(1) make 1000 randomized alternative phenotypes:
perl make_alt_pheno.pl mygwas.fam 1000
(2) run e.g. association on the original GWAS dataset, e.g.
plink --bfile mygwas --assoc --out mygwas_res
(3) run association on all the randomized alternative phenotypes, e.g.
plink --bfile mygwas --pheno mygwas.fam.altpheno.1000.txt --assoc
**NOTE: you can run --assoc or instead --logistic with --covar etc. to run regression with covariates, or --mh to run the Cochrane-Mantel-Haenzel test.
(4) pull out SNP and P-value information:
perl parse_assoc_files.pl
(5) run the SRT on the original dataset:
perl run_SRT_on_ORIGINAL.pl 0.05 mygwas_res.assoc.forSRT
(6) how many SNPs are at the top? (corrects for inflation of p-values):
perl count_sig_SNPs.pl mygwas_res.assoc.forSRT 0.05
(7) run the SRT on the randomized phenotypes (number is the result from (6)):
perl run_SRT_on_SIMS.pl 12075
(8) get the p-values for each pathway:
perl get_SRT_p_value.pl mygwas_res.assoc.forSRT.p0.05.ratios 0.05
See https://sourceforge.net/apps/mediawiki/snpratiotest/index.php?title=Main_Page for more information.
Have fun!
Saturday, May 22, 2010
P-P plots in R
e.g. a plot like this:

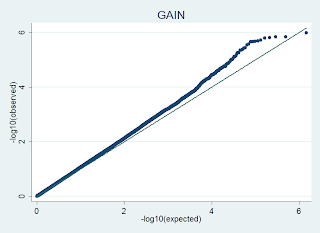
is generated in R using code like this, where the input file has no header, and 1 columns of p-values from a GWAS dataset:
load(sma)
load(plot)
wtccc <- read.delim("wtccc.frequentist.qq", header=FALSE)
wtccclogp <- -log10(wtccc$V1)
wtcccindex <- seq(1,nrow(wtccc))
wtcccuni <- wtcccindex/nrow(wtccc)
wtcccloguni <- -log10(wtcccuni)
qqplot(wtcccloguni,wtccclogp)
plot.qqline(wtcccloguni,wtccclogp)
see http://gettinggeneticsdone.blogspot.com/2009/07/advanced-qq-plots-in-r.html for more.
In STATA, I use code like this, where I generally run an association with the --adjust option in plink, and pull out the observed and expected p-values from the plink output, then where a is column 1 (observed) and b is column 2 (expected):
x = -log10(a)
y = -log10(b)
label if desired: e.g. label variable x "-log10P(obs)"
label if desired: e.g. label variable y "-log10P(exp)"
qqplot x y

is generated in R using code like this, where the input file has no header, and 1 columns of p-values from a GWAS dataset:
load(sma)
load(plot)
wtccc <- read.delim("wtccc.frequentist.qq", header=FALSE)
wtccclogp <- -log10(wtccc$V1)
wtcccindex <- seq(1,nrow(wtccc))
wtcccuni <- wtcccindex/nrow(wtccc)
wtcccloguni <- -log10(wtcccuni)
qqplot(wtcccloguni,wtccclogp)
plot.qqline(wtcccloguni,wtccclogp)
see http://gettinggeneticsdone.blogspot.com/2009/07/advanced-qq-plots-in-r.html for more.
In STATA, I use code like this, where I generally run an association with the --adjust option in plink, and pull out the observed and expected p-values from the plink output, then where a is column 1 (observed) and b is column 2 (expected):
x = -log10(a)
y = -log10(b)
label if desired: e.g. label variable x "-log10P(obs)"
label if desired: e.g. label variable y "-log10P(exp)"
qqplot x y
Friday, May 21, 2010
Kernel Density Plots
Instead of the usual PCA-type plot that looks like this:
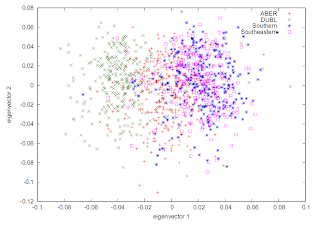
It can be useful to represent this as a Kernel Density plot, like this:
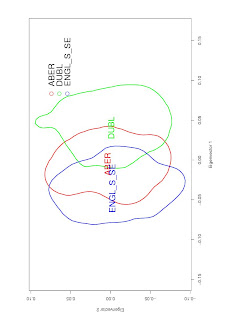
You can typically generate something like this using eigenvectors, e.g. .evec output from EIGENSOFT. The script looks something like this:
## Load library
library(adehabitat)
## Read the eigenvectors from the Eigensoft PCA analysis output file into a variable called, in this case, "ind_pca"
## You may have more columns than just X and Y if you chose to generate more eigenvectors. Adjust accordingly.
ind_pca<-read.table("XXXXX.evec", skip=1, col.names=c("Individual_id", "X", "Y", "A", "B", "C", "D", "E", "F", "G", "H", "Population" ) )
## Extract some useful variables from the table you just loaded
xy<-ind_pca[,c("X","Y")]
id<-ind_pca[,c("Population")]
colours<-c("blue", "green", "red", "orange", "pink", "darkgreen", "purple", "brown", "grey")
means = aggregate(xy, list(id), mean)
## Run the kernel density analysis, this uses the least-square cross-validation (LSCV) and 80% threshhold
hr<-kernelUD(xy, id, h="LSCV")
ver<- getverticeshr(hr, 80)
## Plot the result in a nice pdf
#pdf(file = "Kernel_density_plot.780.eig1_vs_eig2.pdf", width = 15, height = 15)
plot(ver, colborder=colours, colpol="NA", main="Population-wise PC kernel densities based on EIGENSOFT SMARTPCA computed eigenvalues", sub="", xlab="Eigenvector 1", ylab="Eigenvector 2" )
leg.txt<-c("ABER", "DUBL", "ENGL_S_SE")
legend(0.11, 0.08, "ABER", pch=1, col="red", title="", bty="n")
legend(0.11, 0.05, "DUBL", pch=1, col="green", title="", bty="n")
legend(0.11, 0.04, "ENGL_S_SE", pch=1, col="blue", title="", bty="n")
text(means$X, means$Y, names(ver), cex=0.5, col=colours)
#dev.off()

It can be useful to represent this as a Kernel Density plot, like this:

You can typically generate something like this using eigenvectors, e.g. .evec output from EIGENSOFT. The script looks something like this:
## Load library
library(adehabitat)
## Read the eigenvectors from the Eigensoft PCA analysis output file into a variable called, in this case, "ind_pca"
## You may have more columns than just X and Y if you chose to generate more eigenvectors. Adjust accordingly.
ind_pca<-read.table("XXXXX.evec", skip=1, col.names=c("Individual_id", "X", "Y", "A", "B", "C", "D", "E", "F", "G", "H", "Population" ) )
## Extract some useful variables from the table you just loaded
xy<-ind_pca[,c("X","Y")]
id<-ind_pca[,c("Population")]
colours<-c("blue", "green", "red", "orange", "pink", "darkgreen", "purple", "brown", "grey")
means = aggregate(xy, list(id), mean)
## Run the kernel density analysis, this uses the least-square cross-validation (LSCV) and 80% threshhold
hr<-kernelUD(xy, id, h="LSCV")
ver<- getverticeshr(hr, 80)
## Plot the result in a nice pdf
#pdf(file = "Kernel_density_plot.780.eig1_vs_eig2.pdf", width = 15, height = 15)
plot(ver, colborder=colours, colpol="NA", main="Population-wise PC kernel densities based on EIGENSOFT SMARTPCA computed eigenvalues", sub="", xlab="Eigenvector 1", ylab="Eigenvector 2" )
leg.txt<-c("ABER", "DUBL", "ENGL_S_SE")
legend(0.11, 0.08, "ABER", pch=1, col="red", title="", bty="n")
legend(0.11, 0.05, "DUBL", pch=1, col="green", title="", bty="n")
legend(0.11, 0.04, "ENGL_S_SE", pch=1, col="blue", title="", bty="n")
text(means$X, means$Y, names(ver), cex=0.5, col=colours)
#dev.off()
Subscribe to:
Comments (Atom)


